微软研究院近期在人工智能领域迈出了重要一步,于2月20日正式揭晓了其最新研发成果——AI模型BioEmu-1。这款模型专注于预测蛋白质随时间变化的运动轨迹与形态演变,为生物医学、药物研发及结构生物学领域带来了革命性的突破。
蛋白质,这一生命的基本构建单元,在维持生命活动中扮演着不可或缺的角色,从肌肉的形成到疾病的防御,无处不在。科学家们在蛋白质结构研究上已利用深度学习取得了显著成就,能够仅凭氨基酸序列精准预测蛋白质的三维结构。然而,这仅如电影中的一帧定格,未能展现蛋白质这一高度动态分子的全貌。
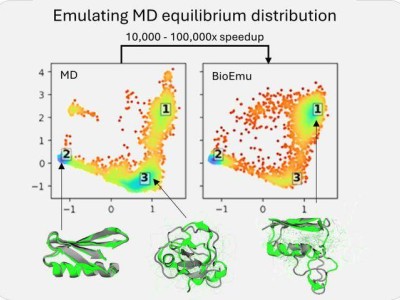
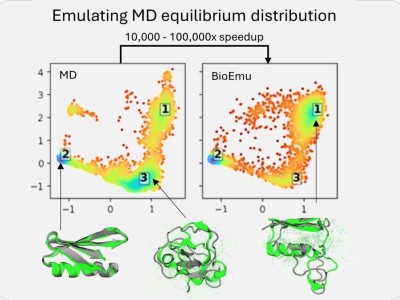
与DeepMind的AlphaFold专注于静态蛋白质结构解析不同,BioEmu-1致力于模拟蛋白质在不同形态间的动态转换过程,为深入理解蛋白质活动机制及设计高效治疗方案提供了强有力的工具。AlphaFold 3虽然在结构生物学上取得了显著进展,特别是在改进蛋白质与其他分子的相互作用模型上,但在预测蛋白质随时间变化的能力上仍有所欠缺。
BioEmu-1正是为了弥补这一空白而生,它能够生成多个可能的蛋白质构象,而非单一的最佳拟合结构,这对于药物研发而言至关重要。该模型采用生成式深度学习技术,从庞大的数据集中学习模式,并结合静态蛋白质结构、分子动力学模拟数据及实验稳定性数据进行训练,从而生成与这些模式相符的新样本。
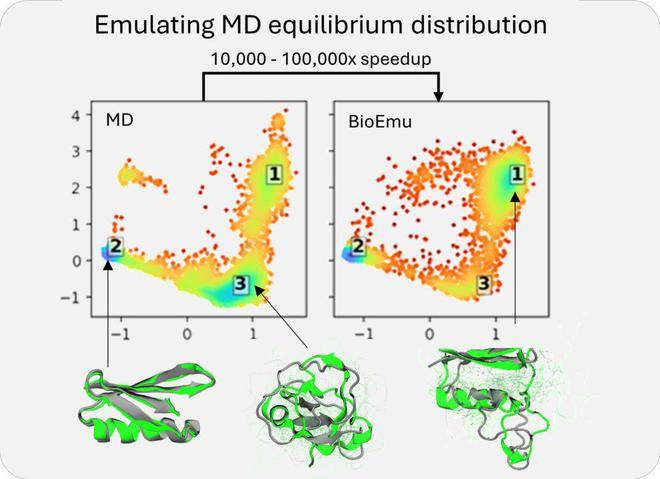
BioEmu-1的核心在于其扩散模型,该模型通过迭代生成蛋白质结构,并根据学习到的约束条件不断提升其准确性。其关键输出包括平衡系综的预测及自由能的预测。为了训练这一模型,微软研究院使用了三种类型的数据集:AlphaFold数据库的结构信息、广泛的分子动力学模拟数据集以及实验性蛋白质折叠稳定性数据集。
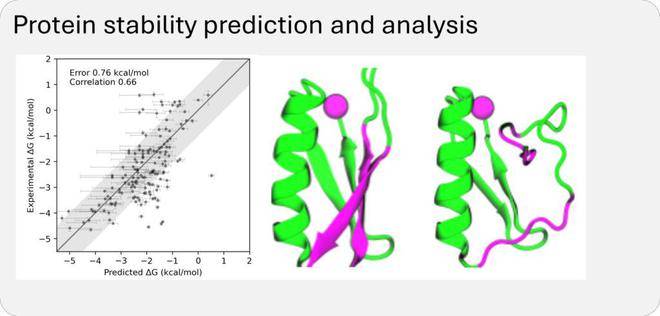
通过这些数据集的训练,BioEmu-1能够识别蛋白质序列与多个不同结构之间的映射关系,预测合理的结构变化,并以正确的概率对折叠与未折叠结构进行采样。这一能力使得BioEmu-1在药物研发领域具有巨大的潜力。
BioEmu-1在效率上也有着显著的优势。它能够每小时生成数千个蛋白质结构样本,相较于传统分子动力学模拟需要数周的时间,BioEmu-1显著加快了研究进程并降低了计算成本。同时,其预测自由能的误差幅度在1 kcal/mol以内,与传统分子动力学模拟相当,但计算成本却大幅降低。
微软研究院的这一创新成果,无疑将为生物医学及药物研发领域带来深远的影响,助力科学家们更深入地理解生命的奥秘,并加速新药的研发进程。